En la Declaración de Helsinki se establece que el proyecto y el método de todo estudio en seres humanos debe describirse claramente en un protocolo de investigación. ...........................................................................
ღEste comité deberá ser independiente del investigador, patrocinador u otro tipo de influencia. ღAdemás los ensayos clínicos deben realizarse siguiendo las normas de buena práctica clínica, obligando así a revisión previa del protocolo por un Comité de ética.
CEI: Órgano independiente y de composición multidisciplinar.
CEIM: Órgano acreditado por el RD 1090/2015.
Sirve para redactar dictámenes en un estudio clínico con medicamentos y productos sanitarios.
FUNCIONES DEL COMITÉ DE ÉTICA DE INVESTIGACIÓN CON MEDICAMENTOS.
A) Evaluar los aspectos metodológicos,éticos y legales y emitir dictamen correspondiente. B) Evaluar las modificaciones sustanciales de los estudios clínicos autorizados y emitir el dictamen correspondiente. C)Realizar un seguimiento del estudio, desde su inicio hasta la recepción del informe final.
COMPOSICIÓN DEL COMITÉ
"El participar en un CEIM será incompatible con cualquier clase de intereses derivados de la fabricación, venta de medicamentos y productos sanitarios"
EL comité de ética se renueva según comunidad autónoma, pero siempre garantizando su continuidad.
DOCUMENTOS Y REQUERIMIENTOS ÉTICOS DE LA INVESTIGACIÓN CIENTÍFICA
¿Cómo hemos llegado a formar nuestros principios éticos biomédicos?
Como sabéis anteriormente no existía ningún reglamento, el cual nos
limitara a experimentar con personas a ton ni son.
Por la crueldad que sufrieron los judíos, personas de raza africana, homosexuales, o cualquier persona que no cumpliera con los "expectativas de ser alemán" en los campos de concentración
y fuera de ellos, es por lo que se ve una necesidad de crear algo que regule los experimentos relacionados con personas, sin que estos supusieran ningún
daño ético y moral para el paciente.
¿Qué paso con los principios éticos después de la segunda guerra mundial?
A principios del
siglo XX existía el derecho a consentir
(defender derechos al paciente)
Código de nuremberg
(1948) recoge una serie de principios que rigen la
experimentación con seres humanos, que resultó de las deliberaciones de
los Juicios de Nuremberg, al final de la Segunda Guerra Mundial,
Informe de Beltmont
fue originado para dar una después ética a los avances de la
investigación biomédica con seres humanos.
Declaración de Ginebra, suiza(1948): es un estatuto
que recoge los deberes éticos para los médicos.
Declaración de Helsinki (1964-2013): ha
sido promulgada por la Asociación Mundial Médica (AMM) como un cuerpo de principios éticos
que deben guiar a la comunidad médica y otras personas que se dedican a la
experimentación con seres humanos. Por muchos es considerada como el documento
más importante en la ética de la investigación con seres humanos.
◊La primera declaración fue realizada por Helsinki (1964 junio)
◊La segunda ha sido sometida a 5 revisiones y 2 clarificaciones, creciendo así, de 11 a 37 párrafos.
◐Es un importante documento, pero no legal.
◐Es un significativo del esfuerzo por autorregularse, por parte de los médicos.
◐Es la base de muchos documentos posteriores
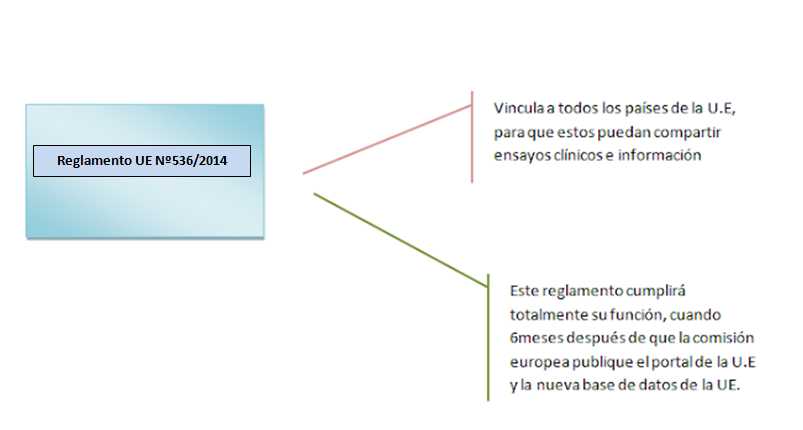
Directiva europea sobre ensayos clínicos 2001-2014:
◊2001: Todos los
pacientes de la U.E, deben seguir está orden.
◊2014: Ya avanzado mucho la
comunicación, compartir información y ensayos clínicos (Que era lo que se más se buscaba)
Convenio sobre derechos
humanos y biomedicina de 1995, del consejo europeo.
Donde se proclama
abiertamente la primicia del interés y bien estar del ser humano
En este convenio se habla
del consentimiento informado, haciendo que, el derecho a la intimidad, bienestar del paciente,
prevalezca sobre el interés del investigador.( Pilar fundamental).
Principios éticos
¿Qué son los principios éticos?¿ Qué incluyen?
Diversos autores han elaborado un modelo moral para quienes trabajan en el ámbito de la salud a fin de proporcionar una referencia practica y conceptual que pueda servir de orientación en situaciones concretas.
Tal modelo, está cimentado en 4 principios:
1. Principio de Beneficiencia:
Se pretende que la actuación del profesional de salud sea beneficiosa
tanto para el personal como para el paciente, siendo este último el más débil,
el que necesita una adecuada y benéfica
ejecución medica.
◊Es decir hacer todo el bien al paciente.
◊Pero tiene un inconveniente: La variedad de conceptos que existen entre
lo que esta bien y lo que esta mal.
2. Principio de No-maleficiencia:
◊Inocuidad de la medicina.
◊Procede de
la ética hipocrática, el ayudar o al menos no dañar.
◊En la praxis hay que evitar hacer daño.
3. Principio de Justicia:
◊Exige tratar a todas las personas con la misma consideración sin hacer
discriminación de raza, ideología o tipo de enfermedad.
◊Esto quiere decir que a misma enfermedad mismo tratamiento o
distribución de recursos.
Principio de Autonomía
Este es el del carácter liberal /radical del
paciente, que en plena posesión de sus facultades mentales y siendo este mayor
de esas, podrá decidir sobre su enfermedad, de tal manera que será solo el que
decide sobre los procedimientos o tratamientos que quiere recibir.
◊Este principio va muy de la mano con el consentimiento informado.
→Donde quedará reflejado de manera clara e
intentando no usar palabras técnicas, todo lo referido al procedimiento o
tratamiento que recibirá el paciente, siendo él, el firmante, como anteriormente
he explicado.
La constitución Española y la investigación científica médica.
Nuestra constitución (1978) a través de leyes importantes establece el
régimen de la libertad de la investigación
En la 1ª: Reconoce y protege el derecho a la producción y creación
científica y técnica (Derecho a crear ciencia).
En la 2ª: Dispone que los poderes públicos promoverán la ciencia y la
investigación, así como técnicas, en beneficio del interés general.
Normativa sobre ensayos clínicos en
la unión europea
✿✿Al iniciar una investigación clínica, es imprescindible reunir información clínica, para preparar la documentación básica relacionada con el estudio.✿✿
MANUAL DEL INVESTIGADOR
"En ella se resumen de manera comprensiva para el investigador las características de preparado a estudiar (Seguridad, Eficacia...). ✿El investigador crea este manual, antes de iniciar el estudio, con ayuda del Farmacéutico o Químico.
Teniendo en cuenta los siguientes aspectos:
Nombre genérico del producto:
◊Denominación internacional. ◊Fórmula química. ◊Descripción cualitativa y cuantitativa de la composición del producto.
Esta información básica del manual está orientada a responder necesidades del investigador sobre recomendaciones,incompatibilidades, referencia sobre dosis, reacciones adversas...además de contar con una estructura.
¤ Procedentes de ensayos clínicos terminados con el preparado en estudio ( si existe) y que hagan referencia a dosis utilizadas, duración del tratamiento, resultado terapéutico y efectos adversos.
Es el conjunto de instrucciones que sirven para determinar la forma en que debe ser desarrollados, a fin, de comprobar la veracidad de la Hipótesis (Eficacia del producto nuevo sobre el anterior).
Es el documento que establece la razón de ser del estudio (Objetivos, diseño, metodología, análisis previo de los resultados, además de las condiciones de trabajo).
◊ Tiene que estar bien estructurado, para otra persona autorizada, pueda leerlo.
◊ Además de estar completo.
◊Elaborado por un equipo multidisciplinar.
◊A propuesta del promotor y con finalidad de conseguir los objetivos establecidos previamente.
◊Es deseable incluir en el protocolo a las personas que con posterioridad van a participar en el ensayo (investigador, farmacéutico del hospital...).
"Es el documento diseñado siguiendo los requisitos establecidos en el protocolo"
"Elemento esencial en el desarrollo de todo ensayo clínico, este documento debe estar en formato simple, para analizar después los datos, siguiendo un orden y además que sean concisos
ESTRUCTURA DE LOS FORMULARIOS DE REGISTRO DE CASOS.
1.Todo tipo de información a registrar: Información que según el protocolo sea necesaria, para valorar eficacia y tolerancia del fármaco en estudio.
2.Forma de cumplimentar las respuestas necesarias para proporcionar dicha información, utilizando para registrar los datos técnicas como la alfanumérica o escalas analógicas.
3. Diseño del gráfico adoptado: Será aquel que facilite que la información se registre e interprete de forma adecuada (orden, instrucciones claras y uniformes).
Reflexión personal
Este tema me ha impresionado. Siempre me ha llamado mucho la atención todo lo relacionado con las enfermedades, los medicamentos y todo su desarrollo
Y gracias ,tanto este tema como el anterior, he podido ir un pasito más allá, sabía que existían los ensayos clínicos, de hecho creía que sabía bastante y a día de hoy he descubierto, que no tenía ni idea.
Ha despertado en mi gran curiosidad por seguir en este mundo, a confirmado mi vocación de buscar curas y seguir pregúntame el porqué de las cosas todos los días.
He visto también como hay una gran conexión entre ensayo clínico y empatía, y me gusta, porque así algún día aparte de encontrar los tratamientos para esas enfermedades, estaremos creando un mundo más empático, una sociedad con corazón, eso que parece que uítimamente se está perdiendo.
En relación a los documentos que hemos tocado en este tema también me ha llamado mucho la atención, ya que , he llegado a palpar lo estricto y sistemático que tienen que ser y además quedando todo reflejando porque verdaderamente juegan con personas y no pueden permitirse cometer ningún fallo más grave de la cuenta. Para mi este ha sido un gran tema y lo finalizó con un deseo.
"Ojalá algún día encontremos un medicamento que cure o alivie la enfermedad de Cronh"





 ღ Este comité deberá ser independiente del investigador, patrocinador u otro tipo de influencia.
ღ Este comité deberá ser independiente del investigador, patrocinador u otro tipo de influencia.








 `
`